|
1 t5 i+ R, U% R: }. u: F- P( x

! ]9 _) e4 ?' y, N- n4 l
摘要
0 W; `1 E/ C1 s+ E 
, G! |5 E* i! Q
随着人为因素对海洋生态系统造成的压力日益增大,对生物多样性进行有效检测的需求也愈发强烈。与基于形态学的检测相比,大宗样品的DNA宏条形码(成本控制更好且耗时更短)分析被越来越广泛的应用在生态系统评估中。但是,在从大量样品中获得原始序列之前,必须做出大量的方法选择。在这里,作者严格审查了用于海洋样品的最新的宏条形码方法,并指出潜在的误差是如何在采样,预处理,DNA提取,marker和引物选择,以及PCR扩增和测序过程中被引入的。在对64项研究完成评估后,作者建议:(1)选择DESS作为样品的固定剂;(2)使用DNeasy PowerSoil试剂盒检测含有痕量沉积物的样品;(3)marker的选择不局限于COI(cytochromeoxidase subunit I;细胞色素氧化亚基I),但最好使用多个marker以获得更高的分类学分辨率;(4)避免TDPCR profiles(touchdownPCR;降落PCR);(5)对每对引物使用固定的退火温度;(6)最少使用3个PCR重复;(7)使用阴性和阳性对照。尽管DNA宏条形码具有大量的技术复杂性,但是作者预计在不久的将来此项技术会有长足的发展,包括长片段测序,可用于分类分配的改进之后的生物信息学以及全基因组信息的使用。
# `9 O1 q1 P8 e/ q+ Y 编译:张政 / j/ ? W0 Z! }
英文标题:
2 i/ J8 e8 K3 F' w Biases in bulk: DNA metabarcoding of marine communities and the methodology involved 8 c7 d, P0 `4 }+ c' \& Z
中文标题: y* Z* S8 Q- v+ Z/ g% d
大量偏差:海洋群落的DNA宏条形码和所涉及的方法论 l3 | J! t) Z" {. ~* }; Y$ j) b3 [
期刊:
3 B. ?9 o# l& ~ Molecular Ecology, 2020
. x' S' R. E; ?4 y 第一作者:
3 y1 q5 _" H5 E5 L H Luna M. van der Loos
) L4 ^1 S8 O1 q% `; E* E& m 通讯作者:
2 ?. W. r& T8 \% _6 t9 j4 M Reindert Nijland
* n+ y+ {4 B/ b% Z- ^1 v; Z& F 作者单位: ) o. \* ?: h Y% j
Marine Animal Ecology group,Wageningen University, P.O. box 338, 6700 AH Wageningen, The Netherlands 1 x) h" A0 f7 M2 ~( v q0 f
Present address: Department of Biology, Phycology Research Group, Ghent University, Ghent, Belgium
1 O* \- W1 E& \, S 前言  / B o$ u% k/ v, ?9 k / B o$ u% k/ v, ?9 k
基于高通量测序技术的DNA宏条形码技术可以同时鉴定同一样品中的许多类群的DNA,它提供了一种省时又经济的选择。基因组DNA可以从大样本(bulk samples;例如:沉积物中分离出或浮游生物网收集到的生物)中提取出来,这些DNA称为群落DNA(community DNA)。另一方面,环境DNA(eDNA)是直接从环境样本中获得的DNA。虽然这两者都称为宏条形码,但是仍然存在很大不同,其中环境样本中的相关DNA相对较少,且容易退化。因此eDNA宏条形码技术主要用于难以直接从环境中获取的生物。近年来,DNA宏条形码技术已经大量应用于海洋生物的多样性调查。此项技术的发展有利于我们跨领域和跨时间的研究比较,但是同时需要统一的标准来规范日常的方法实施。在日常实验中,往往从采样开始就面临多种方法的选择(目标物种,采样周期,样本类型和样本大小)。在整个过程中又有多个阶段需要进一步的方法选择,例如样品固定和分离方法,DNA提取,引物选择以及最后的测序。在整个实验过程中,每个环节都可能引入技术误差,并对结果造成很大影响。因此在这篇文章中,作者主要关注海洋样品DNA宏条形码背后的方法选择,从而对大样本DNA宏条形码技术的偏差和发展瓶颈做出概述。本文主要有以下三个目的:(1)基于全面的文献综述和调查,概述目前使用的方法和大样本DNA宏条形码的潜在偏差;(2)为最佳实践方案和标准程序提供建议;(3)批判性地评估当前的方法瓶颈,并对在现有水平下大样本DNA宏条形码的未来发展提供意见。
, o1 k) K, D: H( U& v 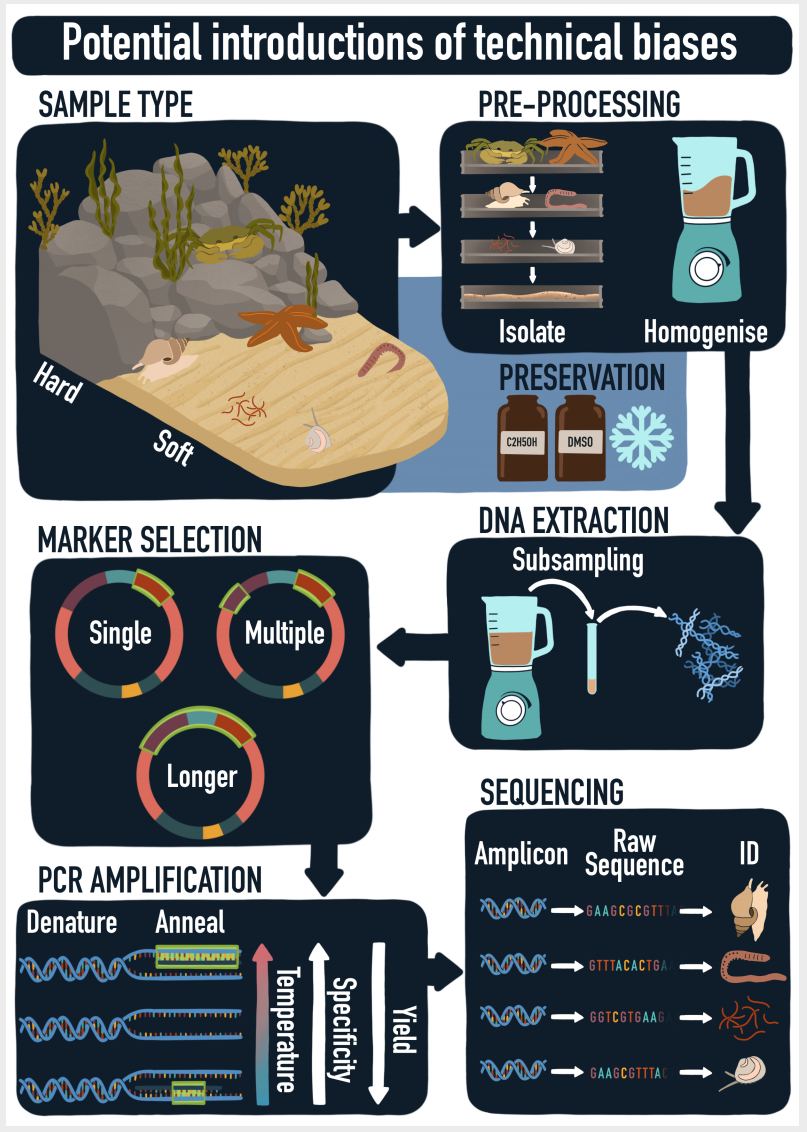
( I" Q* H" B0 F9 e 图1 从原始样品到原始序列和分类分配过程的示意图。在采样,预处理,DNA提取,marker选择,PCR扩增和测序过程中可能会引入技术偏差。 ) \# X5 b/ R1 M6 W+ f% L$ o9 d
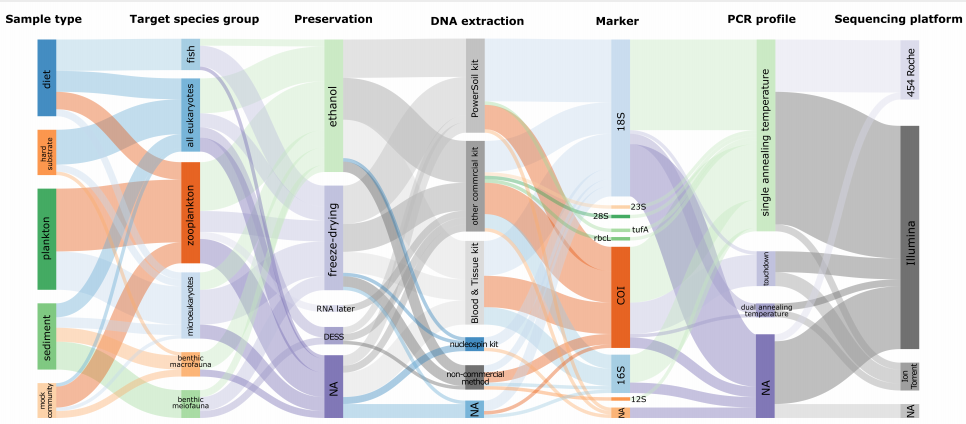
- ]2 d+ d8 f4 E 图2 对2010年至2019年发表的64项研究的bulk DNA宏条形码的七个阶段使用方法的相对频率,包括样品类型、目标物种、保存、DNA提取、marker、PCR和测序平台。线条的粗细与使用这种方法的文章数量相对应。“NA”表示没有报告或不需要相应阶段/方法的研究。 8 O; x s' p/ v5 J6 ?+ k( u. _
结果和讨论
$ [* M* @& a- n7 q; q4 U* _4 c 大样本DNA宏条形码技术对食物样本(分粪便或胃内容物)和浮游生物样本效果明显,因为这些样本中的生物往往难以识别。在研究设计中需要提前明确自己的目标物种,从而针对不同的物种选择不同的研究手法。样品中的DNA质量会从采样之后迅速下降,尤其是在高温环境下。因此样品必须在采集之后迅速进行预处理或者在现场妥善保存。目前大多数研究选择96-100%乙醇对样品进行保存,尤其是针对浮游生物样品。沉积物样品大多数选择冷冻的方法保存,而DESS(含有EDTA的盐饱和DMSO缓冲液)则针对小型动物和线虫的研究。Yoder等人研究发现,DESS能够保证足够的DNA和较为完整的形态学保存,其保存效果要好于乙醇。同时众多研究表明DESS保存的样品具有更高质量的DNA和更多的PCR产物,并将其推荐为首选样品保存方法。
+ a+ f, I; K V" ~ 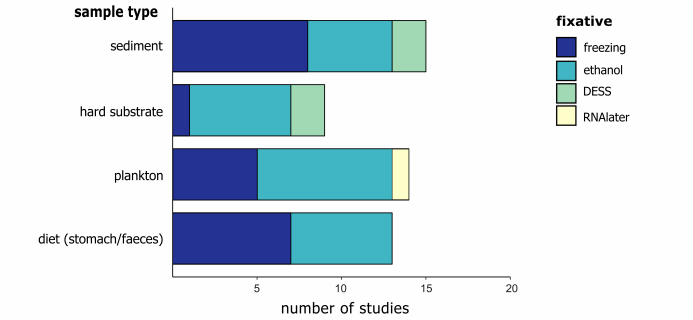
" Z+ M1 Z+ x& s) c* o
图3 针对不同样本类型保存方法 ' a1 h; k/ n' ^
由于大块样品会存在不均质的现象,因此我们一般不会使用原始样品进行DNA提取,而是先对样品进行必要的预处理,例如有机物和无机物的分离。对于沉积物来说,里面存在的一般抑制剂会影响PCR反应中酶的活性,因此这就需要进行必要的物质分离。即使从有机组分中将无机物分离出来,样品往往仍然是异质的,而如果我们要检测所有的物种,这就需要对整个样品进行DNA提取,但是由于需要回收的生物量太大,从而无法实现这个目标。我们可以选择将样本进行充分的均质化,使用子样本来代替整体样本进行DNA提取。假设充分均质的子样本中包含所有的物种DNA,然而较大和较丰富的物种在均质子样本中贡献了更多的物种组织,这可能导致后续的检测结果向这些物种倾斜,而较小和较稀有的物种可能仍未检测到。这一问题在多样性多和物种更复杂的样本中更为突出。Elbrencht等人提出,在DNA提取前要根据研究目标对样本进行必要的尺寸分流,特别是样本中的物种大小相差多个数量级时。
' [/ f8 b; h& G# [ 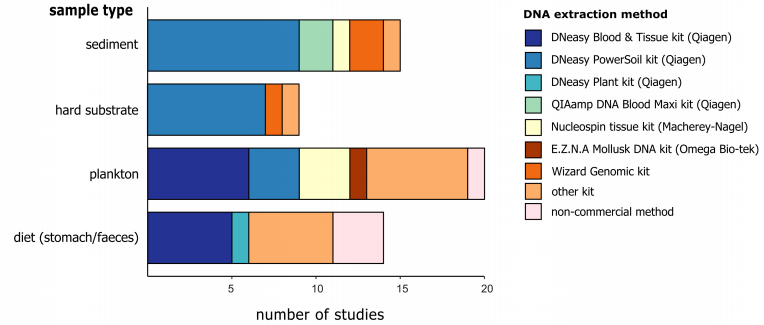
- }; O- b8 I! l 图4 针对不同样本类型的DNA提取方法 ( U% \3 k9 B- U8 [5 G
作者通过文献调研发现,DNeasy PowerSoil试剂盒是目前用于沉积物研究最常用的试剂盒,其可以有效获得高纯度的DNA并且去除沉积物中含有的抑制剂。在进行DNA宏条形码实验时,对DNA marker区域的选择同样是一个关键因素。18S标记与12S和16S一样,它比COI更加保守,更容易设计引物,但是在物种分类上的能力较弱。在选择barcode或者引物的时候需要在覆盖度和特异性(分类学的分辨率)上寻找平衡。调研发现,当研究的目标生物为较小的底栖生物时,多选择18S的V1-V2或者是V9区域,而研究一般真核生物时,多使用18S的V4区域。研究发现由于物种的分辨率较低,18S通常会低估物种数量,例如Clarke等人发现,COI和18S在门水平上显示出了相似的覆盖度,但是在物种水平上,COI的解释能力时18S的3倍多。 $ f( j: d; v/ X( T
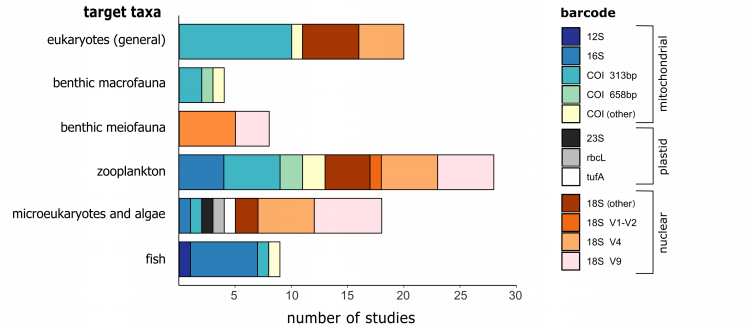
. O9 W0 o, w- q/ o# s 图5 针对不同研究目标的引物选择 越来越多的研究表明,使用多对引物将会得到更可靠的物种丰度,然而现在的研究绝大多数仍然在使用单一引物,这是因为使用多对引物会成倍增加工作量和成本。但是Zhang等人在研究中指出,为了达到覆盖度和特异性的最佳平衡,需要使用多个引物或者多个引物组合的标记。测序的最后两个步骤——PCR扩增和测序本身——都是引入偏差的重要来源。在这些过程中,DNA分子是随机选择的,这个随机性对稀有序列的检测会产生特别大的影响,从而影响数据集的再现性。要选择适合的退火温度,温度过低会导致某个产物的放大,而温度过高又会降低产量。调研发现18S的V9和V1-V2区主要使用57℃的退火温度,而18S的V4区主要使用50℃。最佳退火温度不仅取决于引物的GC含量,还取决于引物浓度和所使用的DNA聚合酶类型。为了避免退火问题或者在处理低质量DNA时,PCR的循环数通常为30以上。虽然提高PCR循环数不会影响由于聚合酶错误而引起的突变数量,但是会增加形成的嵌合体数量或放大误差。考虑到PCR扩增的随机性,强烈推荐在进行大样品(bulk)宏条形码中使用至少3个PCR重复。但是调研中发现,48%的研究没有使用PCR重复,因为增加PCR重复会增加成本和实验时间,因此具体的PCR重复数选择需要根据特定的研究问题或研究目标而决定。如果你的目标是检测稀有物种,增加测序深度比增加PCR重复数更重要。当你的目标是获得尽可能完整生物多样性信息,或者是某些reads相对数量变化的结论非常重要的时候,使用至少3次PCR重复可能会有比较好的结果。作者指出,在测序深度较低的时候,使用MisSeq可能比Illumina遗漏更多的生物多样性。通过提高测序深度来增加检测的多样性,对于DNA含量较低或多样性较高的样品尤为重要。与广泛使用的Illumina测序平台相比,nanopore测序能够测序任何长度的DNA分子,它可以提供更好的分辨率,更长的片段以供分析。调研的9家欧洲研究所中有5家在测序过程中使用了PCR重复,8家使用Illumina测序平台,一家使用OxfordNanopore测序手段,但是有两家目前正在尝试使用这个平台。; T: F" n3 o" O% [1 V' f) y( a
其实最重要的一点是在测序过程中使用阴性和阳性对照。这两种对照将有助于PCR的质量监控(阴性对照:监控质量的同时不会损失生物样本,不会影响测序深度;阳性对照:在扩增和文库制备过程中检查和纠正样品之间的交叉污染)
) l2 B; n0 a, h# E& Z4 ~8 N 建议: % b/ j T( H" z- [& |; Z
1. 在采样之前要明确目标样本,根据样本选择相应的方法。 ! x+ z& X( j1 u! C! F
2. 如果有染色观察任务,在染色之前要分离样本。
# j# J# _6 T" j6 y! b# \! t5 D4 T& R. ] 3. 在进行再次取样和DNA提取之前,必须对样本进行混匀。 / K: w/ C1 a3 J& Y/ |7 A4 I
4. DESS是首选的样本保存溶液,它可以产生最高数量和质量的DNA,易于在野外使用。 $ n3 m* E- Y& c4 @
5. 推荐使用Qiagen FNeasy PowerSoil试剂盒 + A% c0 F6 ~# ~9 H# z3 v$ t
6. 如果研究的目标是发现一个系统中尽可能多的生物多样性时,强烈建议使用多个marker/引物 2 V2 n f2 [6 _: X8 {# C
7.  CR操作方案一定要严谨,这个过程极易引入误差 CR操作方案一定要严谨,这个过程极易引入误差 6 V6 y2 v! q N% a' ]( z; Z
8. 固定退火温度
7 }/ O4 y( V( E$ g 9. CR循环数最好小于35 . h5 |% F: g% q9 j: q
10. 是否使用PCR重复要根据实验目的和科学问题来选择,以下几种情况建议使用至少3个PCR重复
' |( h! x8 |, Z2 s# @. k# V 1)目的是为了找到尽可能完整的生物多样性 . T6 m* t) j P: r \4 d; o( G
2)分析时会考虑reads的相对数量的变化
0 D+ f' p! X: M5 o0 j: a 3)在操作时可能会降解或减少目标DNA ; d4 [2 j* D' o2 G* t& L D! C
11. 在以下情况不使用PCR重复 % m; B" x2 M5 S0 F+ \: n* ~. Q, r: v
1)样本可以提供大量你的目标物种的遗传信息 - U: Q' p( W) b i. m
2) 分析的时候考虑存在—缺失数据框架(presence-absence data frames) # y$ [$ d9 F; e0 V6 U* G
12. 引入几个阴性对照和至少1-2个阳性对照。阳性样本最好来自不同的生态系统,避免样本之间的交叉污染。
0 S" I# ], o- P. p" e+ \5 W6 X 挑战和期望
3 H. r3 Z9 o+ N1 V1 W- _% p) V 1. 与传统形态学的匹配 由于目前的检测方案和生态状况评估主要基于传统的形态学观察测,因此构建宏条形码与历史观测之间的桥梁显得无比重要。目前有许多检测工作涉及大尺度和长期的形态学观测,如果这些项目突然转变成宏条形码将势必会导致观测到的物种组成发生变化。因此,只有当用宏条形码得到的结果与用传统形态分类学得到的结果相匹配时,才考虑采用DNA宏条形码。目前宏条形码技术在海洋领域大范围使用,其获得的分类单元的数量在大多数情况下是要多于形态学观测的结果的,但是宏条形码结果不能涵盖所有的传统形态学观测的结果。传统形态学观测可能会受到分类学分配、寄生物种以及肉眼难以观测的物种等问题的影响,但是该方法仍然可以对传统的检测任务产生较好的结果。而宏条形码的优势在于可以检测到肉眼无法观测的细小物种,同时其结果可以覆盖大范围的物种分类水平。但是宏条形码技术受限于数据库的完备和准确水平。
, v( }9 R4 m2 b- \- H 2. 定量相关 Reads的比例与样本中原始物种的比例缺乏相关性。目前有关这两者之间的相关性的研究也逐渐成为热点。虽然有些研究发现reads的比例和物种丰度之间存在良好的相关性,但是随着数据的转换,这些相关性会随之消失。Lamb等人认为reads丰度与生物量之间弱相关关系,与个体数量没有相关性。能够量化物种丰度是未来物种检测的一项要求。8 p: Z* E; D0 p! @" i# d
3. 未来期望
2 _* I+ \9 A3 \5 o0 i, ~+ m# \ 1) 改进原始序列处理和获取分类信息的生物信息学手段 这篇综述集中在从生物样本中获得原始序列的方法过程中可能引入的误差。然而,在获得原始序列后,利用生物信息学方法进行分类分配也会对群落产生影响。要选择合适的分类学信息学处理方法和阈值,阈值过于保守会去除稀有物种,反之会导致多样性膨胀。目前ASV的使用在逐年增加,它能够更精准的分析遗传变异6 W/ c( R) V& W& |( h* q$ ~
2)改进相关数据库和marker 参考数据库的大规模扩展,不仅有越来越多的物种被纳入其中,同时还希望加入完整的线粒体基因组和全场核糖体序列,为不是短短的单个标记基因。2 H! D! x& T& t% v! L
3)去除PCR步骤 PCR过程中引入的误差可能比其他过程引入的要多得多。无扩增的线粒体DNA或核糖体基因直接测序或许可以解决这个问题。
" y: r" ^, V i$ u- W 4)使用基因组数据 目前,测序技术和数据处理的成本效益已经足够高,可以省略特定的扩增或富集,而只是直接对样本中的DNA进行测序。同样,为了实现有效的分类分配,参考数据库的质量将是主要的瓶颈。基于基因组skims的鸟枪宏基因组方法值得一试(Peel et al 2019)。1 L. @ y! U, J, s1 }
4. 宏条形码到底是一种独立的还是互补的技术? 3 ?3 J) b5 d+ R1 L& @6 x) a l
说了那么多,我们可能忽略了形态学观测也会存在误差。根据研究目的的选择性抽样本身就会引入误差,宏条形码技术可以在另一个较大尺度上帮我们思考物种多样性和丰富度。同时,形态学观测和宏条形码之间的一对一比较本身就是不合理的,因为“物种”这个概念往往不能单纯的从字面上理解,同时二者都提供了基于研究目的的可靠的数据。因此如何评价宏条形码技术要基于你的研究目的,它既可以单独的作为研究主体又可以作为传统观测的补充。 & K; b) g# Z, b! @# A

2 s7 T% f J- a: L, x3 U
参考文献:
* `6 V+ Y2 ]9 B' E Peel N, Dicks LV, Clark MD,Heavens D, Percival‐Alwyn L,Cooper C et al (2019). Semi‐quantitative characterisation of mixed pollen samples using MinIONsequencing and Reverse Metagenomics (RevMet). Methods in Ecology and Evolution 10: 1690-1701. 6 L ^8 c& o6 ~
van der Loos LM, Nijland R(2020). Biases in bulk: DNA metabarcoding of marine communities and themethodology involved. Mol Ecol. 5 K, {7 ]7 W: @9 @, w
原文链接: 4 q4 \; u1 o$ L0 S5 [1 X3 r' ]1 V
https://onlinelibrary.wiley.com/journal/1365294x # P Y# R7 v3 O! w! U: [6 c7 P6 ~" M
 
+ ~9 s( b3 l2 |8 X- c
中国科学院生态环境研究中心
/ d5 Z. p! H7 M1 ` v% @ i; X' E" C+ q- k9 H- s9 u6 z; h, R" S- `
环境生物技术重点实验室 ) B, ~0 @8 j# l: ?+ A' p, v' Z0 D% o
邓晔 研究员课题组发布
: b5 Y/ U# \, _! [. ? 编译:张政
# \! g' N5 n+ Q0 H5 W) E1 L 关注微生态笔记~    - `# c% D( d% g
- `# c% D( d% g 哎呀! 听说“阅读原文”
3 n/ H9 y; j; {- _/ `, a2 q 可以读文献 可是我点不到呢
7 b1 F v) Y7 Z9 @2 ]
+ Z, G+ k& L5 K& H/ q5 R5 Q4 u) Z7 Y z+ B8 }
; R, C" J% k, d- m, Z7 q, ]
/ U/ Y1 {, C1 H5 B: _) ?" d | 

