|
7 g( O' ~0 S$ v8 T0 f
注:为保证全方位的展示文章的精华,此文篇幅略长,请广大读者耐心看完,绝对良心推荐! . _, G! A1 i6 Z5 S0 \
全球海洋DNA病毒宏观和微观多样性 8 F9 j2 c! j g# m
Marine DNA Viral Macro- and Microdiversity from Pole to Pole * p- {+ D9 I' t
作者:Ann C. Gregory, Ahmed A. Zayed, Na´ dia Conceic¸ a˜ o-Neto, et al. 8 ?; T0 M9 y- O, ?: X
期刊:Cell C: Y6 I9 I" M8 G
时间:2019-5
3 o3 d! F: f, L2 M* q; h7 ]( j* M 影响因子:31.4
9 r* x$ ^! V6 t5 S9 U) l 推荐指数:★★★★★ 8 i4 y N" m. u- i
一、研究背景 - C+ s0 G6 g/ V0 T! d
微生物驱动着大部分生态系统,而病毒影响着微生物的生命周期、基因流动和代谢产出。由于缺乏关于病毒分类的数据和参考基因组,目前仍然难以评估生态系统水平对病毒类群多样性的影响。在此,我们建立了一个比之前规模约大12倍、拥有195728种全球海洋DNA病毒组的数据库,该数据库包括来自北冰洋、有离散基因簇的病毒类群。全球海洋可分为5个生态区,其中2个在北极。在地区及全球范围内,分析了在宏观(群体多样性)和微观(群落内部遗传变异)水平上影响病毒类群多样性的模式和驱动因子。这些模式有时与宏观生物的模式类似,表明温热带海洋浅表水层和北冰洋是生物多样性的热点地区,并提出了解释这些现象的机制假说。在总结生态系统模式时,对海洋病毒的深层次理解显得至关重要。
! J/ [2 }& }8 ~% Q9 l4 v 二、实验设计 ( D N w4 T# C9 ^6 A
1、样品采集
- V1 [: l: ]3 }5 Q 在北冰洋及其附近海域的20个采样点共收集到41个样品。提取样品DNA,构建文库,使用HiSeq 2000进行测序(101 bp,双端reads)。需要注意的是DNA的提取和建库针对的都是<0.2 μm的ds DNA病毒。 5 f: N# A9 `, A- `) _3 X6 s2 U' ]
2、样品处理 2 M7 T$ s+ i& b2 J# ?; G
在Tara和Malaspina 2个海域采集和保存样品的方法都是一样的,但后者有更长的reads长度(分别为101 bp和151 bp)。本实验用r2值、p-值和贝叶斯信息准则验证了生态变量和实验方法差异对分析结果的影响。结果表明,造成样本之间差异的主要是生态和类群结构,而非实验方法。 - X9 E- V# @+ b, |/ v( b8 ]! ^/ i
3、定量和统计学分析 4 `5 K. i: a; ~6 l* e5 Y
对所有样品单独进行组装。在组装前对GOV 2.0中的Malaspina样本进行数据质控。筛选≥1.5kb的contigs,去除比对上其他物种的污染基因组序列。总共鉴定到848507种病毒contigs,在GOV 2.0数据库中找到488130种病毒类群。
$ }* {4 j" P3 c' x# p 用R语言分析病毒宏观α多样性(Shannon’s H)和β多样性(Bray-Curtis dissimilarity)。选择在GOV 2.0据库中至少在1个样品中出现过、70% contig的平均测序深度≥10×的病毒类群,分析其微观多样性。在各数据库中对翻译成蛋白的序列进行注释分析。 0 Y- h" k6 Q1 A5 n4 E& P# K% @
对5个地区的病毒群体进行多地区性、区域性和地区性划分,根据生物地理学划分出ARC-H(高多样性)和ARC-L(低多样性)两个地区;对GOV和GOV 2.0两个数据库进行比较;对前人发表的16S OUT数据进行α分析,其分析结果与病毒多样性进行关联分析。此外,文章还分析了海岸远近、海洋深度和季节对病毒类群的影响。 ; q, m/ u. S8 Z7 ^4 b& J% @; a9 L
三、实验结果
7 v; G# P6 t1 X( X+ O; A; l 1、数据库信息 . L+ A) |' t4 q: Q* P) t& U
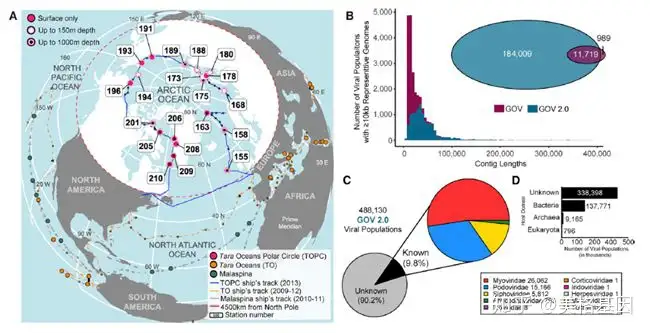
全球海洋病毒组 (GOV 2.0) 数据库是在GOV的基础上,补充了来自海洋中层水域(150 m-1000 m)样品的测序结果和深度组装信息,目前GOV 2.0有145个来自全球海洋样品的3.95 Tb的序列信息(图 1A)。另外我们还增加了在Tara海洋极圈采集到的41个新样品,它们代表受气候影响最大的海洋地区和极端环境下的北冰洋病毒组。 5 q" q$ L2 |4 a
分析病毒生物多样性的第一步是从测序序列中鉴定出疑似病毒的contigs并具体归类到种群中(contigs≥10 kb,且超过70%的contigs的平均核酸相似性(average nucleotide identity, ANI)≥95%)。通过以上流程,在GOV 2.0中共鉴定出195728种病毒类群,是GOV的12倍之多。GOV中有92%的contigs在GOV 2.0中都可以找到,而后者的序列长度(44 kb)是前者(18 kb)的2.4倍(图 1B)。新增的180448种病毒类群中有58%是在GOV样品的基础上,通过进一步组装和深度测序的情况下得到,而剩下的42%来自41个北冰洋病毒组。通过新方法(STAR Methods)对短contigs(5-10 kb)进行分析,鉴定出292402种病毒contigs、 488130种病毒类群,其中90%病毒类群无法分类到目前已知的病毒科当中,剩下的10%主要是dsDNA病毒科和噬菌体(图 1C, 1D)。 ' K" a2 u4 V* [! @" e
图1 全球海洋基因组2.0(GOV 2.0)2 、病毒类群的“界限” ! r ~ {2 t5 z H/ _5 h6 l
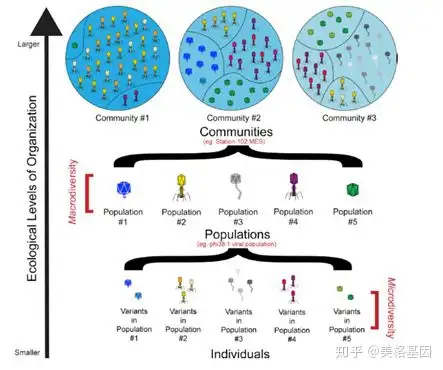
目前,在种水平上对原核生物和真核生物的定义仍存在争议,而对病毒来说更是如此。由于基因流动性太强,现在还没有足够的数据能在种水平上对病毒进行分类,因此我们引入类群(population)的概念。文章还对用≥95% ANI这一阀域来划分病毒类群的普遍性进行了评估。在允许“局部”核苷酸相似性低至18%的情况下,将宏基因组reads回贴到COV 2.0的488130个病毒类群上。结果表明核酸相似性<92%的reads无法比对回基因组序列(图 2B),而≥95% ANI这一阀域几乎适用于所有的GOV2.0病毒类群(99.9%),包括短contigs的病毒类群。这意味着数据库中大部分病毒类群都存在序列离散特性。事实上,所有通过宏基因组组装的dsDNA病毒类群都形成了离散的基因簇,并且可以用≥95%全基因组ANI这一阀域来界定。
- Z1 F9 e1 ?2 \- p2 z 图2 GOV 2.0病毒类群存在离散的类群界限3、宏类群分析揭示5个生态区特性
+ R! Z# G. l$ V 把整个病毒序列划分为离散且有生物学意义的类群后,我们尝试利用宏基因组得到的丰度信息,建立跨全球海洋、跨多生态层级的病毒类群多样性的模式和驱动因子。经多方法评估,可将来自GOV 2.0的145个病毒群体划分为5个宏群体,亦表示5个生态区。这5个自然生态区分别是北极圈(ARC)、南极洲(ANT)、深海区(BATHY)、温热带浅海(TT-EPI)和中深海(TT-MES)。 # g3 y) `- m9 k7 |& @$ `3 n \
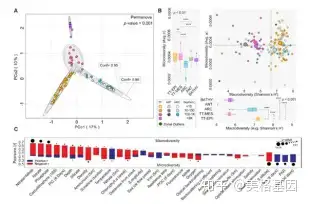
海洋微生物种群的理化结构也许是影响病毒类群结构的最重要因素,因此我们对病毒生态区的预测因子和驱动因子进行了单维度(图5 A)和多维度(图5B)评估。与全球微生物研究相似,温度是塑造这些生态区的最重要因素,同样也在病毒-宿主互作方面起到重要作用。
' x1 b# L, a1 P, R. d$ ~9 j+ d 图3 组织的自然生态水平在每个生态区中寻找特殊的病毒适应因子时发现,每个样品中至少有124882个基因是正向选择的,其中82%的基因无法得到功能注释,其余的18%主要是结构或DNA代谢相关基因。据此推测,宿主给海洋病毒类群造成了强大的选择压力。 / O. w# S6 ^5 k3 m5 K
4、各生态区中(间)的病毒宏观和微观多样性和潜在驱动因子
. e1 |$ I8 J$ s: V# C+ n 为探索各生态区的病毒多样性模式,我们对每个样品的宏观差异和整个类群的微观差异进行了计算。在区水平上,病毒宏观和微观多样性表现的主要是负相关趋势。有研究证明,造成这种负相关的原因是,强的微观多样性通过加强竞争性排斥来降低宏观多样性。
% o) P; I. r- F S/ P1 X; v 但在样品层级上,即使是在同一个生态区内,病毒类群的宏观和微观多样性并不相关。据此推测这是地域性驱动因子造成的(图 4C)。在上层水域,营养物质和病毒宏观多样性呈强烈的负相关,而光合成有效辐射(PAR)与病毒微观多样性呈强烈的正相关。 ( d$ Q% Y3 F6 I0 P
图4 病毒类群划分为五个具有不同宏观和微观多样性水平的生态区以上结果表明,在病毒宏观多样性水平上,藻华宿主多样性降低会造成病毒的秩丰度曲线偏移,使得与藻华相关的病毒群体丰度增加并倾向于占绝对优势。虽然在Tara海洋远征中我们并没有以藻华为研究目标,但病毒宏观多样性与叶绿素a(图5C)及无机碳微粒浓度为负相关(图4C)。在上层水域,PAR是病毒微观多样性潜在的主要驱动因子。高强度的PAT可能会对病毒的宿主群体产生短期效应的影响,进而为病毒产生新的生态位,增强微观多样性,促进现存病毒类群发生变异。 3 v2 m% k" Y; O( j. J
图5 全球病毒宏观多样性的生态驱动因子分析5、与传统生态区梯度相悖的病毒宏观和微观多样性及潜在的驱动因子
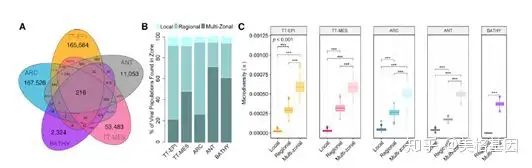
+ Y2 P2 P2 K- E' M* L 本文探索了各生态区中(间)的病毒多样性和地理分布的关系(图 6A),展示了在不同生态区中优势病毒的地理分布差异。如多区域病毒类群主要分布在ANT和BATHY(图 6B),而特定区域病毒类群主要分布在TT-EPI和ATC,在TT-MES中区域病毒类群和特定区域病毒类群数目几乎相同(图 6B)。而在所有的生态区中,病毒类群的微观多样性都随着病毒地理范围的扩大而增加(图 6C)。
4 H+ O/ ]. `8 q" _! t 图6 地理分布范围与病毒微观多样性呈正相关关系在陆地和海洋环境中,绝大部分动植物群落都表现出纬度多样性,而病毒宏观和微观多样性都是随着纬度多样性梯度变化而变化的,除了多样性都增加的ARC区域(图 7A)。在赤道线的印度洋和大西洋,高丰度的病毒宏观和微观多样性是一致的,而在太平洋的除外(图 7B、7C)。在北冰洋,病毒不仅有丰富的多样性,而且还表现出特有的模式。水体营养受气候影响(图 7D)的2个特殊区域分别为高(ARC-H)和低(ARC-L)多样性区,这2个区的宏观和微观多样性都具有显著差异(图 7E)。 4 G: Q3 i M, a( S
有文献报道,微生物和病毒多样性随着海洋深度的增加而降低,但我们的研究结果表明,病毒的宏观多样性在海洋浅表最高并随着海洋深度的增加而降低,微观多样性在水深小于200 m的时候没有降低,在水深大于200 m时急剧增加。(图 7F)。据此推测区域特殊细菌种群宏观多样性的增加会影响病毒微观多样性,病毒微观多样性的增加会引起细菌种群宏观多样性的减少。 3 {: @, l* ]0 w1 V/ g
图7 病毒宏观和微观生物多样性的全球趋势四、小结与亮点 ; _0 x, i) b- U
1、将世界海洋划分出5个不同于其他有机体生物地理模式的生态区。
, b( M0 R8 Z7 M2 |1 q9 W. T 2、系统的研究各生态区海洋病毒的宏观和微观多样性及其驱动因子。 $ B+ G$ q6 \$ {3 I
3、 上层海洋水域和北冰洋成为研究病毒生物多样性的热点地区。
# q; E2 ]7 X* r4 N 总之,本研究对Tara海洋生态系统进行了全方位的测量,为将病毒纳入基因-生态系统模型提供基础,对指导人们做出关乎人类和地球发展的海洋生态系统管理决策具有重要意义。
9 N7 I2 R: U( S. b 您可能还喜欢:
& d4 j5 m% w8 Y7 f - [$ S5 F- n8 d. g7 P O
5 b3 C9 z! K7 K; U, J
0 `& s, e, [5 F7 N0 l+ Y! r, @" |2 `0 F& e4 Y- U' N
0 [- ?% l) V$ I3 k5 L0 g+ m) g0 r4 X, v: m p
! K- m) A3 @+ K0 ]" I ^- N( P1 s
|